Meer gedetailleerde informatie
Het gen alfa-1-antitrypsine. Het gen dat codeert voor het a1-antitrypsine (a1AT; ook wel a1-proteinase inhibitor of a1-PI genoemd) is gelokaliseerd op chromosoom 14. Het heeft een lengte van 12.2 kb en bestaat uit zeven exonen. Het a1AT is een lid van de familie van serine protease inhibitors, die “serpins” worden genoemd, en waarvan de genen deels zijn geclusterd op chromosoom 14 op locatie 14q32.1. Tot de familie van serpins behoren o.a. ook het a1-antichymotrypsine, a2-antiplasmine, antithrombine en C1-inhibitor.
Het gen is zeer polymorf, en in de Europese populatie zijn meer dan 75 verschillende allelen beschreven. De meest voorkomende mutaties in het a1AT gen zijn de Z en S mutaties, waarbij er wel een eiwit wordt gevormd. De minder vaak voorkomende Null-mutatie heeft als gevolg dat er geen eiwit wordt gevormd.
Daarnaast zijn polymorfismen beschreven in een enhancer sequentie in het a1AT gen die betrokken is bij de regulatie van de expressie van a1AT.
Het eiwit
1AT is een acuut fase eiwit van 394 aminozuren (52 kDa). Deficiëntie van a1AT en de associatie met longemfyseem werd voor het eerst beschreven in 1963. Het eiwit werkt als enzymremmer van enzymen (o.a. elastase en proteinase 3) die worden uitgescheiden door neutrofiele granulocyten. Door het roken van sigaretten wordt een ontstekingsreactie in de longen met veel granulocyten onderhouden. Gezien de rol die neutrofielen spelen in de pathogenese van emfyseem, wordt verondersteld dat een tekort aan a1AT kan resulteren in een enzymatische afbraak van de alveolaire septa die een kenmerk is van de pathologie van longemfyseem.
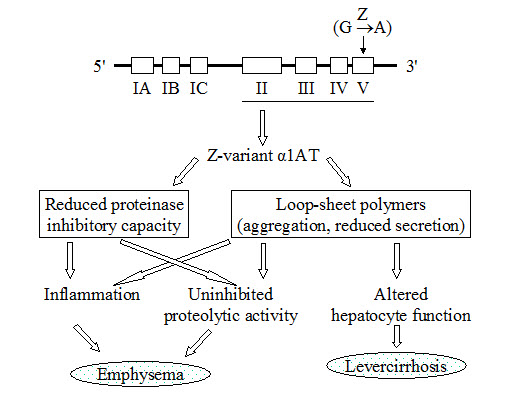
Als eiwit is a1-AT op een karakteristieke manier opgevouwen. Het enzym-bindende centrum van a1-AT steekt zodanig uit dat gesproken wordt van een geëxposeerde mobiele loop. Deze loop wordt opengeknipt na binding aan een enzym als elastase waarna het complex van a1-AT en enzym zich in een andere vorm opvouwt, met als resultaat dat het enzym is geïnactiveerd. De Z mutatie (een G naar A mutatie) leidt ertoe dat een negatief geladen glutamaat residu wordt vervangen door een positief geladen lysine.
De Z mutatie is gelokaliseerd aan de basis van de loop, en leidt ertoe dat de loop zich spontaan nestelt in een b-sheet van een tweede a1AT molecuul. Dit leidt tot de vorming van de kenmerkende loop-sheet polymeren, die accumuleren in het endoplasmatisch reticulum van de levercellen, een proces dat de basis vormt voor de ontwikkeling van levercirrhose die soms wordt waargenomen bij patiënten met de Z mutatie. De S mutatie leidt ertoe dat op positie 264 een glutamaat residu wordt vervangen door een valine. Deze mutatie heeft als gevolg dat het S-eiwit gevoeliger is voor intracellulaire afbraak.
De cel
De belangrijkste bron van synthese van het a1AT is de lever, terwijl lokaal ook macrofagen en epitheelcellen in alveoli kunnen bijdragen aan de synthese. Het gevolg van de Z mutatie is vorming van a1AT polymeren die accumuleren in de levercel. Hierdoor wordt er weinig a1AT aan plasma afgegeven, met als gevolg een deficiëntie in o.a. de long, en een beschadiging van de levercel.
Recent onderzoek heeft aangetoond dat polymeren van a1-AT, die ook in longsecreten zijn aangetoond, neutrofiele granulocyten kunnen aantrekken en stimuleren, en zo aan de lokale ontsteking en ontwikkeling van longemfyseem kunnen bijdragen.
De populatie
De Z mutatie is waarschijnlijk vanuit Scandinavië door de Noormannen naar West- en Zuid-Europa gebracht. Door verdere emigratie naar het Noord-Amerikaanse continent is de aandoening daar, naast cystic fibrosis, een veel voorkomende erfelijke afwijking. In Scandinavië is de frequentie van het type Z homozygoot 1 : 1600. In de rest van Europa, inclusief Nederland, is de frequentie van ZZ individuen in de algehele populatie ten westen van de lijn Barcelona, Milaan, Wenen en Leningrad geschat op 1 : 3500, waarbij er subpopulaties zijn met een frequentie van 1 : 1500.
Diagnostiek
Bij screening volstaat een bepaling van het serumgehalte van a1AT, dat bij de Z mutatie 10 % is van de hoeveelheid die wordt gevonden bij personen met het normale M allel. Hierbij dient men zich te realiseren dat de concentraties van a1AT bij een patiënt kunnen variëren omdat a1AT een acuut fase eiwit is. Is de a1AT concentratie verlaagd, dan wordt aanbevolen om een fenotypering te bepalen d.m.v. iso-electrische focussering. Daarnaast is genotypering ook mogelijk, maar dit is kostbaarder.
Door de WHO wordt aanbevolen om alle individuen die voor hun 40ste levensjaar onbegrepen dyspneuklachten hebben te screenen op de deficiëntie. Voorts wordt aanbevolen om bij iedere nieuwe casus ook broers en zusters te onderzoeken omdat minstens 25% kans bestaat dat bij één van hen ook de deficiëntie aanwezig is en in zo’n geval het advies te stoppen met roken als een zinvolle interventie beschouwd mag worden.